1 Constructing metacells
In this chapter, we will demonstrate metacell construction using three different methods: SuperCell in R, MetaCell-2 (MC2) and SEACells in Pyhton.
For this, we will first use a dataset of bone marrow cells from the SeuratData package. This dataset contains around 30K cells and is an example of a dataset with well defined cell types. For an example of more continuous data, see chapter 4.2.
1.1 SuperCell (R)
In this section, we construct metacells using the R package SuperCell.
1.1.1 Method
The SuperCell method first reduces the gene expression space using principal component analysis (PCA) and computes euclidean distances based on the reduced space. Using the euclidean distances, a single-cell kNN graph is built and metacells are identified by applying the walktrap community detection algorithm. The number of metacells obtained can be chosen by the user by defining the graining level parameter.
The code provided in this section is adapted from the author’s github documentation. For more information on the method, please refer to our review (Review?) and the original paper (Bilous et al. 2022).
Importing R packages
To run SuperCell, the following R package needs to be imported:
if(system.file(package='SuperCell') == ""){
remotes::install_github("GfellerLab/SuperCell", force = TRUE, upgrade = "never")
}
library(SuperCell)
library(Seurat)
#> The legacy packages maptools, rgdal, and rgeos, underpinning this package
#> will retire shortly. Please refer to R-spatial evolution reports on
#> https://r-spatial.org/r/2023/05/15/evolution4.html for details.
#> This package is now running under evolution status 0
#> Attaching SeuratObject
# If you have Seurat V5 installed, specify that you want to analyze Seurat V4 objects
if(packageVersion("Seurat") >= 5) {options(Seurat.object.assay.version = "v4"); print("you are using seurat v5 with assay option v4")}1.1.2 Data loading
We will run SuperCell on a single-cell dataset composed of around 30’000 bone marrow cells. Please follow the section 0.2 to retrieve these data from the SeuratData package and save the data in the following file: “data/bmcite/singlecell_seurat_filtered.rds”.
MC_tool = "SuperCell"
proj_name <- "bmcite"
annotation_label <- "celltype_simplified"
cell_types <- c("Unconventional T", "Naive T cell", "Non-Naive CD4 cell", "CD14 Mono", "B cell", "Non-Naive CD8 cell",
"NK", "GMP", "CD16 Mono", "pDC", "cDC2", "Prog_B", "Plasmablast", "HSC", "LMPP", "Prog_DC", "MEP")
celltype_colors <- c("#1E88E5", "#FFC107", "#004D40", "#9E9D24",
"#F06292", "#546E7A", "#D4E157", "#76FF03",
"#26A69A", "#AB47BC", "#D81B60", "#42A5F5",
"#2E7D32", "#FFA726", "#5E35B1", "#EF5350","#6D4C41")
names(celltype_colors) <- cell_types
sc_data = readRDS(paste0("data/", proj_name, "/singlecell_seurat_filtered.rds"))1.1.3 Filtering steps
In this tutorial, the data have been pre-filtered and SuperCell does not require further filtering steps.
1.1.4 Building metacells
Metacells construction using SuperCell requires one main input, i.e. a matrix of log-normalized gene expression data which will be used to compute PCA to subsequently build a knn graph for metacells identification.
Important optional inputs are:
(i) the graining level (gamma parameter),
(ii) the number of neighbors to consider for the knn graph (k.knn parameter),
(iii) the number of principal components to use to generate the knn graph (n.pc parameter),
and (iv) the number of most variable genes to consider for PCA (n.var.genes parameter).
Data pre-processing
SuperCell builds its knn graph based on Euclidean distances defined in the PCA space.
PCA computation is performed on the log-normalized gene expression data in the SCimplify SuperCell function.
In the following code chunk, we use Seurat to normalize and visualize the data:
sc_data <- NormalizeData(sc_data, normalization.method = "LogNormalize")
sc_data <- FindVariableFeatures(sc_data, nfeatures = 2000)
sc_data <- ScaleData(sc_data)
#> Centering and scaling data matrix
sc_data <- RunPCA(sc_data, npcs = 50, verbose = F)
sc_data <- RunUMAP(sc_data, reduction = "pca", dims = c(1:30), n.neighbors = 30, verbose = F)
#> Warning: The default method for RunUMAP has changed from calling Python UMAP via reticulate to the R-native UWOT using the cosine metric
#> To use Python UMAP via reticulate, set umap.method to 'umap-learn' and metric to 'correlation'
#> This message will be shown once per session
UMAPPlot(sc_data, group.by = annotation_label, cols = celltype_colors)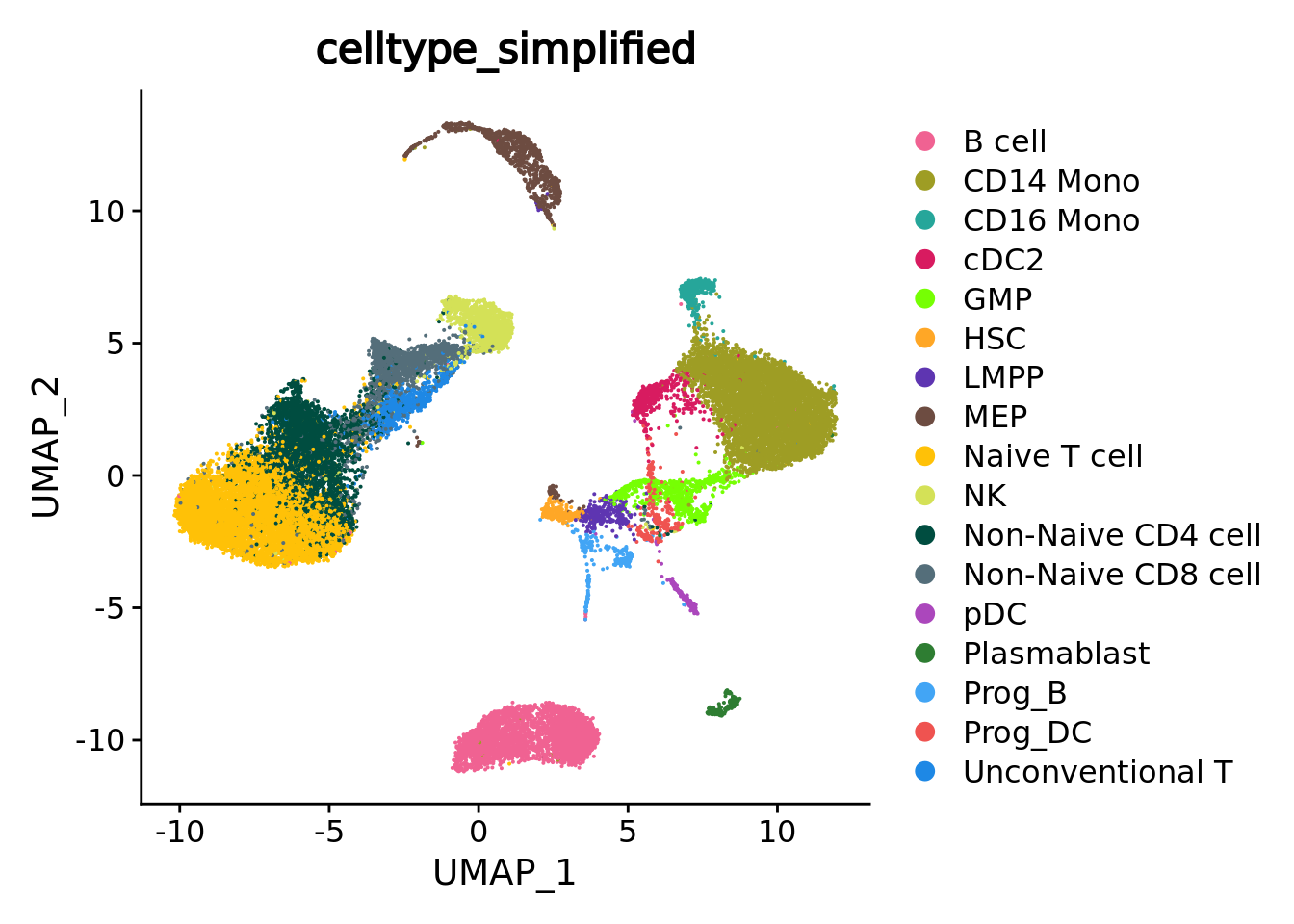
Setting up SuperCell parameters
In this tutorial, we will run SuperCell using the 30 first principal components resulting from the PCA. We chose a graining level of 25 and a number of neighbors of 15 for the knn step.
gamma = 50 # the requested graining level.
k_knn = 30 # the number of neighbors considered to build the knn network.
nb_var_genes = 2000 # number of the top variable genes to use for dimensionality reduction
nb_pc = 50 # the number of principal components to use. Metacells identification
The metacells are identified using the SCimplify function from the SuperCell package.
MC <- SuperCell::SCimplify(Seurat::GetAssayData(sc_data, slot = "data"), # single-cell log-normalized gene expression data
k.knn = k_knn,
gamma = gamma,
# n.var.genes = nb_var_genes,
n.pc = nb_pc,
genes.use = Seurat::VariableFeatures(sc_data)
)SCimplify returns a list containing the following main elements:
(i) the single-cell assignments to metacells (membership),
(ii) the metacell sizes (supercell_size),
(iii) the single-cell graph (graph.singlecell),
(iv) the metacell graph (graph.supercells),
(v) the genes used for metacell identification (genes.use).
Retrieve aggregated metacell data
The supercell_GE() function can be used to generate a metacell counts matrix (aggregation of gene expression across all cells belonging to each metacell).
Two modes can be used for single-cell aggregation, i.e. averaging of log-normalized gene expression or summing up raw counts (using the mode parameter).
Note that we provide raw counts for the aggregation in this tutorial to match the aggregation steps using PC2 and SEAcells (see 1.2 and 1.3).
Data normalization will thus be needed for downstream analyses on the metacell counts matrix.
MC.GE <- supercell_GE(Seurat::GetAssayData(sc_data, slot = "counts"),
MC$membership,
mode = "sum"
)
dim(MC.GE)
#> [1] 17009 6131.1.5 Annotate metacells (using available annotations)
We can assign each metacell to a particular annotation using the supercell_assign() function.
By default, this function assigns each metacell to a cluster with the largest Jaccard coefficient to avoid biases towards very rare or very abundant clusters.
Alternatively, assignment can be performed using relative (method = "relative", may cause biases towards very small populations) or absolute (method = "absolute", may cause biases towards large populations) abundance.
print(annotation_label)
#> [1] "celltype_simplified"
MC$annotation <- supercell_assign(clusters = sc_data@meta.data[, annotation_label], # single-cell annotation
supercell_membership = MC$membership, # single-cell assignment to metacells
method = "absolute"
)
head(MC$annotation)
#> 1 2 3
#> "Non-Naive CD8 cell" "Non-Naive CD8 cell" "LMPP"
#> 4 5 6
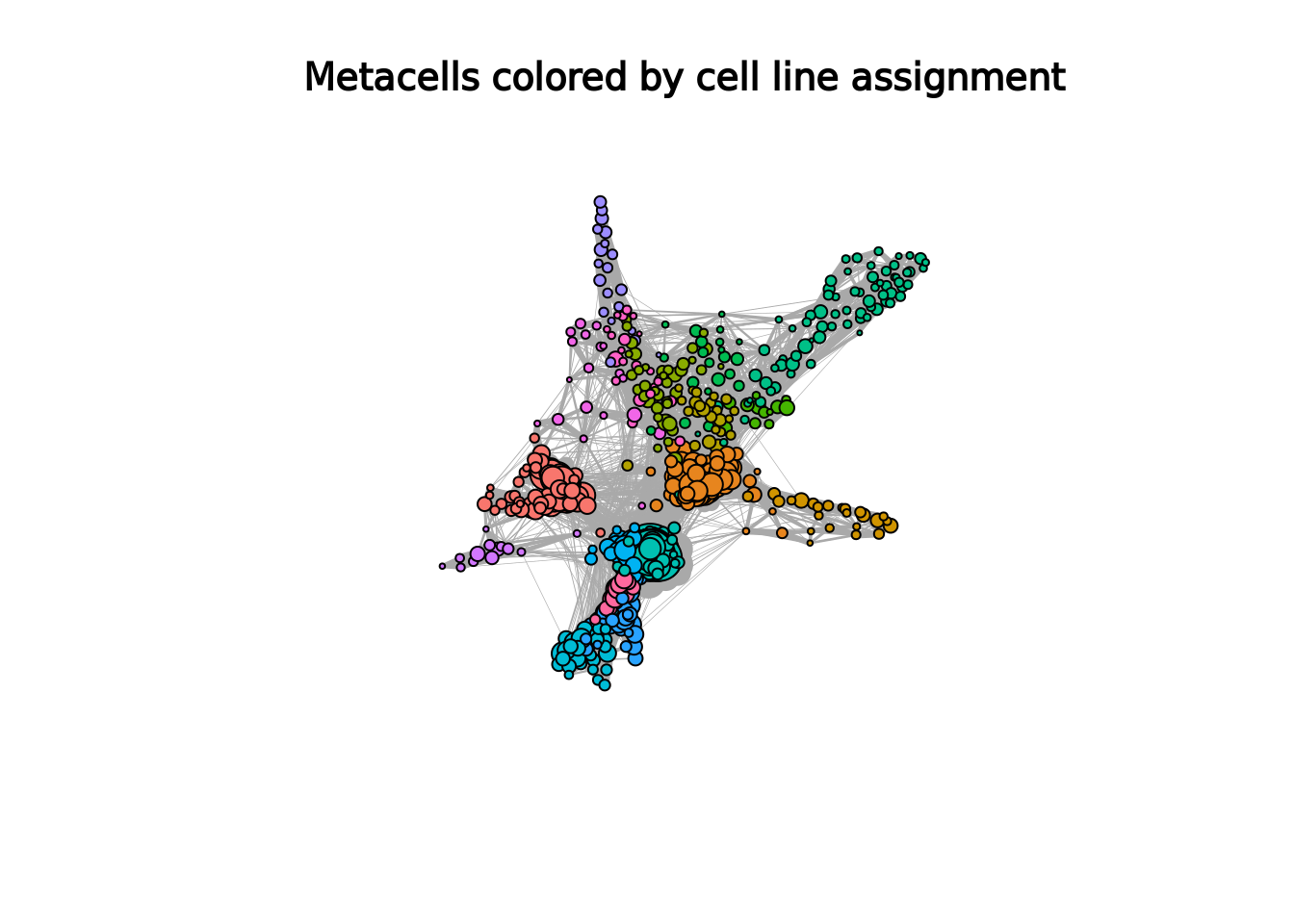
#> "B cell" "CD14 Mono" "Non-Naive CD4 cell"The SuperCell package provides the supercell_plot function to visualize the metacell network (igraph object where number of nodes corresponds to number of metacells),
which is stored in the MC list in graph.supercells.
The metacells can be colored with respect to a vector of annotation.
supercell_plot(
MC$graph.supercells,
group = MC$annotation,
seed = 1,
alpha = -pi/2,
main = "Metacells colored by cell line assignment"
)
1.1.6 Save output
For future downstream analyses in R (section 3.1), metacell counts can be saved in a Seurat object. Here we also save in the Seurat object the PCA components and genes used in SCimplify for future QC analysis (See 2).
colnames(MC.GE) <- as.character(1:ncol(MC.GE))
MC.seurat <- CreateSeuratObject(counts = MC.GE,
meta.data = data.frame(size = as.vector(table(MC$membership)))
)
MC.seurat[[annotation_label]] <- MC$annotation
# save single-cell membership to metacells in the MC.seurat object
MC.seurat@misc$cell_membership <- data.frame(row.names = names(MC$membership), membership = MC$membership)
MC.seurat@misc$var_features <- MC$genes.use
# Save the PCA components and genes used in SCimplify
PCA.res <- irlba::irlba(scale(Matrix::t(sc_data@assays$RNA@data[MC$genes.use, ])), nv = nb_pc)
pca.x <- PCA.res$u %*% diag(PCA.res$d)
rownames(pca.x) <- colnames(sc_data@assays$RNA@data)
MC.seurat@misc$sc.pca <- CreateDimReducObject(
embeddings = pca.x,
loadings = PCA.res$v,
key = "PC_",
assay = "RNA"
)
if(packageVersion("Seurat") >= 5) {
MC.seurat[["RNA"]] <- as(object = MC.seurat[["RNA"]], Class = "Assay")
}
print(paste0("Saving metacell object for the ", proj_name, " dataset using ", MC_tool))
#> [1] "Saving metacell object for the bmcite dataset using SuperCell"
saveRDS(MC.seurat, file = paste0('./data/', proj_name, '/metacell_', MC_tool,'.rds'))We can also use the supercell_2_Seurat() function from the SuperCell package.
This function takes as inputs the metacell count matrix (output of the SuperCell supercell_GE() function) and the output of the SuperCell SCimplify() function
to output a Seurat object containing normalized metacells gene expression data as well as the first (N.comp) principal components of PCA performed internally using user defined set of genes (by default the genes used for metacells constructions).
MC.seurat <- supercell_2_Seurat(
SC.GE = MC.GE,
SC = MC,
fields = c("annotation", "supercell_size"), # elements of MC to save as metacell metadata
var.genes = MC$genes.use,
N.comp = 10
)
if(packageVersion("Seurat") >= 5) {
MC.seurat[["RNA"]] <- as(object = MC.seurat[["RNA"]], Class = "Assay")
}
saveRDS(MC.seurat, file = paste0('./data/', proj_name, '/metacell_', MC_tool,'.rds'))For future downstream analyses in python (section 3.2), metacell counts can be saved in an Anndata object:
MC.seurat.ad <- anndata::AnnData(
X = Matrix::t(Seurat::GetAssayData(MC.seurat, slot = "counts")),
obs = MC.seurat@meta.data
)
anndata::write_h5ad(anndata = MC.seurat.ad, filename = paste0('./data/', proj_name, '/metacell_', MC_tool,'.h5ad'))#> used (Mb) gc trigger (Mb) max used (Mb)
#> Ncells 3494496 186.7 11654105 622.4 14567631 778.0
#> Vcells 177948554 1357.7 438523727 3345.7 438519104 3345.71.2 MC2 (Python)
In this section, we construct metacells using Metacell-2 (MC2) implemented in Python.
1.2.1 Method
Metacell-2 (MC2) is a python tool to construct metacells and is the updated version of the MetaCell algorithm, which introduced the concept of metacell. MC2 applies a two-phase divide-and-conquer approach. Cells are randomly divided into piles of ~10k cells and initial metacells are built applying a MetaCell-like approach per pile, i.e. based on a single-cell kNN graph built from log-normalized counts using a set of highly variable genes. Then, transcriptionally similar metacells are grouped into metagroup piles for the identification of final metacells and outliers identification. Note that prior to metacell identification, the MC2 framework recommends gene filtering steps. The choice of the genes used by the method is of high importance to assure good quality of the metacells.
The code provided in this section is adapted from the author’s tutorial. For more information on the method, please refer to our review (Review?) and the original paper (Ben-Kiki et al. 2022).
Importing python packages
To run MC2, the following python packages need to be imported:
import os
import numpy as np
import pandas as pd
import anndata as ad
import scanpy as sc
import matplotlib.pyplot as plt
import seaborn as sns
import metacells as mcIf you don’t have these packages installed, please refer to the section 0.1.
1.2.2 Data loading
We will run Metacell-2 (MC2) on a single-cell dataset composed of around 30’000 bone marrow cells. Please follow the section 0.2 to retrieve these data from the SeuratData package and save the data in the following file: “data/bmcite/singlecell_anndata_filtered.h5ad”.
MC_tool = "MC2"
proj_name = "bmcite"
annotation_label = 'celltype_simplified'
ad = sc.read(os.path.join("data", proj_name, "singlecell_anndata_filtered.h5ad"))
ad.var.index = ad.var.genesWe initialize the name of the anndata (in the unstructured annotations) object using the mc.ut.set_name() function from the MC2 package.
mc.ut.set_name(ad, proj_name)1.2.3 Filtering steps
MC2 requires that standard filtering steps such as doublet filtering are performed outside of the MC2 framework. In addition to standard data filtering steps, the MC2 package proposes functions to filter the single-cell data at the gene and at the cell level (See author’s vignette). At the gene level, the filtering steps consist in excluding genes based on biological knowledge (e.g. mitochrondrial genes) as well as based on their expression levels. The latter genes include genes with zero expression or low expression levels and “bursty lonely genes” (i.e., genes with high expression levels but no correlation with any other gene). At the cell level, filtering is performed based on cells UMI counts.
Gene filtering
In the following code chunk, we exclude genes using the mc.pl.exclude_genes()function from the MC2 package.
Based on the authors vignette, we consider a minimal list of genes to exclude, i.e., sex-specific and non-coding genes as well as the mitochondrial genes.
To complete this list of genes, an iterative approach can be used following the guidelines of the MC2 authors in a second vignette.
The mc.pl.exclude_genes() function will filter out:
i) the known-to-be-excluded genes defined by the user as gene names or gene names patterns (EXCLUDED_GENE_NAMES and EXCLUDED_GENE_PATTERNS parameters respectively),
and ii) the “bursty lonely genes”.
EXCLUDED_GENE_NAMES = ["XIST", "MALAT1", "NEAT1"]
EXCLUDED_GENE_PATTERNS = ['MT-.*']
mc.pl.exclude_genes(
ad,
excluded_gene_names=EXCLUDED_GENE_NAMES,
excluded_gene_patterns=EXCLUDED_GENE_PATTERNS,
random_seed=123456
)
#> set bmcite.var[bursty_lonely_gene]: 1 true (0.005879%) out of 17009 bools
#> set bmcite.var[properly_sampled_gene]: 17009 true (100%) out of 17009 bools
#> set bmcite.var[excluded_gene]: 17 true (0.09995%) out of 17009 boolsCell filtering based on UMIs counts
In the MC2 framework, cells with very low and very high UMI content will be filtered out (PROPERLY_SAMPLED_MIN_CELL_TOTAL, PROPERLY_SAMPLED_MAX_CELL_TOTAL variables defining thresholds in the next code chunk).
Also, cell filtering based on UMI counts in excluded genes is also performed(PROPERLY_SAMPLED_MAX_EXCLUDED_GENES_FRACTION variable).
Since our dataset has been pre-filtered, very lenient cutoffs will be used in this tutorial.
The following code chunk defines these parameters.
To adapt them to your datasets, we advise you to explore the distributions of total UMI counts and UMI counts in excluded genes, as recommended and described in the MC2 original vignette.
PROPERLY_SAMPLED_MIN_CELL_TOTAL = 200
PROPERLY_SAMPLED_MAX_CELL_TOTAL = 10000
PROPERLY_SAMPLED_MAX_EXCLUDED_GENES_FRACTION = 0.25The number of UMIs in excluded genes is computed using the mc.tl.compute_excluded_gene_umis() function and cells are filtered out using the mc.pl.exclude_cells() function.
Additional cells can be filtered out by adding a cell description columns in the obs data frame in the anndata oject. This annotation should be a boolean indicating whether the cell should filtered out or not.
The name of this column should be provided to the mc.pl.exclude_cells() function via the additional_cells_masks parameter.
mc.tl.compute_excluded_gene_umis(ad)
mc.pl.exclude_cells(
ad,
properly_sampled_min_cell_total=PROPERLY_SAMPLED_MIN_CELL_TOTAL,
properly_sampled_max_cell_total=PROPERLY_SAMPLED_MAX_CELL_TOTAL,
properly_sampled_max_excluded_genes_fraction=PROPERLY_SAMPLED_MAX_EXCLUDED_GENES_FRACTION # ,
# additional_cells_masks=["|doublet_cell"]
)
#> set bmcite.obs[properly_sampled_cell]: 29581 true (96.44%) out of 30672 bools
#> set bmcite.obs[excluded_cell]: 1091 true (3.557%) out of 30672 boolsAfter performing the two-step filtering (genes and cells), the “cleaned” data can be extracted using the mc.pl.extract_clean_data() function.
# Extract clean dataset (with filtered cells and genes)
ad = mc.pl.extract_clean_data(ad)
#> set bmcite.clean.obs[full_cell_index]: 29581 int32s
#> set bmcite.clean.var[full_gene_index]: 16992 int32s1.2.4 Building metacells
Defining lateral genes
To build metacells, we need to define lateral genes, which are genes with strong biological signal which is independent of cell-state, e.g. cell-cycle genes.
These genes will be ignored for computing cells similarity and building metacells
but will be considered to define outlier cells (i.e., expression levels of lateral genes should be consistent within metacells).
In the following chunk, we consider a minimal list of lateral genes (provided by the MC2 authors) including cell-cycle and ribosomal genes and
mark them in the MC2 object using the mc.pl.mark_lateral_genes() function.
LATERAL_GENE_NAMES = [
"ACSM3", "ANP32B", "APOE", "AURKA", "B2M", "BIRC5", "BTG2", "CALM1", "CD63", "CD69", "CDK4",
"CENPF", "CENPU", "CENPW", "CH17-373J23.1", "CKS1B", "CKS2", "COX4I1", "CXCR4", "DNAJB1",
"DONSON", "DUSP1", "DUT", "EEF1A1", "EEF1B2", "EIF3E", "EMP3", "FKBP4", "FOS", "FOSB", "FTH1",
"G0S2", "GGH", "GLTSCR2", "GMNN", "GNB2L1", "GPR183", "H2AFZ", "H3F3B", "HBM", "HIST1H1C",
"HIST1H2AC", "HIST1H2BG", "HIST1H4C", "HLA-A", "HLA-B", "HLA-C", "HLA-DMA", "HLA-DMB",
"HLA-DPA1", "HLA-DPB1", "HLA-DQA1", "HLA-DQB1", "HLA-DRA", "HLA-DRB1", "HLA-E", "HLA-F", "HMGA1",
"HMGB1", "HMGB2", "HMGB3", "HMGN2", "HNRNPAB", "HSP90AA1", "HSP90AB1", "HSPA1A", "HSPA1B",
"HSPA6", "HSPD1", "HSPE1", "HSPH1", "ID2", "IER2", "IGHA1", "IGHA2", "IGHD", "IGHG1", "IGHG2",
"IGHG3", "IGHG4", "IGHM", "IGKC", "IGKV1-12", "IGKV1-39", "IGKV1-5", "IGKV3-15", "IGKV4-1",
"IGLC2", "IGLC3", "IGLC6", "IGLC7", "IGLL1", "IGLL5", "IGLV2-34", "JUN", "JUNB", "KIAA0101",
"LEPROTL1", "LGALS1", "LINC01206", "LTB", "MCM3", "MCM4", "MCM7", "MKI67", "MT2A", "MYL12A",
"MYL6", "NASP", "NFKBIA", "NUSAP1", "PA2G4", "PCNA", "PDLIM1", "PLK3", "PPP1R15A", "PTMA",
"PTTG1", "RAN", "RANBP1", "RGCC", "RGS1", "RGS2", "RGS3", "RP11-1143G9.4", "RP11-160E2.6",
"RP11-53B5.1", "RP11-620J15.3", "RP5-1025A1.3", "RP5-1171I10.5", "RPS10", "RPS10-NUDT3", "RPS11",
"RPS12", "RPS13", "RPS14", "RPS15", "RPS15A", "RPS16", "RPS17", "RPS18", "RPS19", "RPS19BP1",
"RPS2", "RPS20", "RPS21", "RPS23", "RPS24", "RPS25", "RPS26", "RPS27", "RPS27A", "RPS27L",
"RPS28", "RPS29", "RPS3", "RPS3A", "RPS4X", "RPS4Y1", "RPS4Y2", "RPS5", "RPS6", "RPS6KA1",
"RPS6KA2", "RPS6KA2-AS1", "RPS6KA3", "RPS6KA4", "RPS6KA5", "RPS6KA6", "RPS6KB1", "RPS6KB2",
"RPS6KC1", "RPS6KL1", "RPS7", "RPS8", "RPS9", "RPSA", "RRM2", "SMC4", "SRGN", "SRSF7", "STMN1",
"TK1", "TMSB4X", "TOP2A", "TPX2", "TSC22D3", "TUBA1A", "TUBA1B", "TUBB", "TUBB4B", "TXN", "TYMS",
"UBA52", "UBC", "UBE2C", "UHRF1", "YBX1", "YPEL5", "ZFP36", "ZWINT"
]
LATERAL_GENE_PATTERNS = ["RP[LS].*"] # Ribosomal
# This will mark as "lateral_gene" any genes that match the above, if they exist in the clean dataset.
mc.pl.mark_lateral_genes(
ad,
lateral_gene_names=LATERAL_GENE_NAMES,
lateral_gene_patterns=LATERAL_GENE_PATTERNS,
)
#> set bmcite.clean.var[lateral_gene]: 245 true (1.442%) out of 16992 boolsSome genes have higher variances than expected which could lead to false positive outlier identification.
Users can mark those genes as noisy genes using the mc.pl.mark_noisy_genes() function.
NOISY_GENE_NAMES = [
"CCL3", "CCL4", "CCL5", "CXCL8", "DUSP1", "FOS", "G0S2", "HBB", "HIST1H4C", "IER2", "IGKC",
"IGLC2", "JUN", "JUNB", "KLRB1", "MT2A", "RPS26", "RPS4Y1", "TRBC1", "TUBA1B", "TUBB"
]
# This will mark as "noisy_gene" any genes that match the above, if they exist in the clean dataset.
mc.pl.mark_noisy_genes(ad, noisy_gene_names=NOISY_GENE_NAMES)
#> set bmcite.clean.var[noisy_gene]: 20 true (0.1177%) out of 16992 boolsTo extend this list of lateral genes, users can use the relate_to_lateral_genes function to identify genes that are highly correlated with the predefined lateral genes.
The use of this function is described in the vignette from the MC2 authors.
Define target_metacell_size (graining level)
By default, MC2 will build metacells with a size of 96 cells per metacells.
Users can vary the target_metacell_size parameter to reach a desired graining level.
gamma = 75
target_metacell_size = gammaMetacells identification using the divide and conquer approach
The construction of metacells by MC2 is performed using the mc.pl.divide_and_conquer_pipeline() function.
Note that by default all cores of the system will be used for the metacells construction.
To change this behavior and adapt the number of cores the MC2 authors propose to use the mc.pl.guess_max_parallel_piles() and mc.pl.set_max_parallel_piles() functions
to adapt the number of processed in parallel depending on the available memory.
The mc.pl.divide_and_conquer_pipeline() function associates each cell to a metacell or defines the cell as outlier.
These assignments are found in the obs layer of the anndata object.
max_parallel_piles = mc.pl.guess_max_parallel_piles(ad)
mc.pl.set_max_parallel_piles(max_parallel_piles)
mc.pl.divide_and_conquer_pipeline(
ad,
target_metacell_size = target_metacell_size,
random_seed = 123456)
#> set bmcite.clean.var[selected_gene]: * -> False
#> set bmcite.clean.var[rare_gene]: 10 true (0.05885%) out of 16992 bools
#> set bmcite.clean.var[rare_gene_module]: 16982 outliers (99.94%) and 10 grouped (0.05885%) out of 16992 int32 elements with 1 groups with mean size 10
#> set bmcite.clean.obs[cells_rare_gene_module]: 29562 outliers (99.94%) and 19 grouped (0.06423%) out of 29581 int32 elements with 1 groups with mean size 19
#> set bmcite.clean.obs[rare_cell]: 19 true (0.06423%) out of 29581 bools
#> set bmcite.clean.var[selected_gene]: 1807 true (10.63%) out of 16992 bools
#> set bmcite.clean.obs[metacell]: 29581 int32s
#> set bmcite.clean.obs[dissolved]: 5 true (0.0169%) out of 29581 bools
#> set bmcite.clean.obs[metacell_level]: 29581 int32s
ad.obs.metacell.head
#> <bound method NDFrame.head of a_AAACCTGAGCTTATCG-1 382
#> a_AAACCTGAGGTGGGTT-1 148
#> a_AAACCTGAGTACATGA-1 286
#> a_AAACCTGCAAACCTAC-1 84
#> a_AAACCTGCAAGGTGTG-1 202
#> ...
#> b_TTTGTCATCCGAGCCA-1 389
#> b_TTTGTCATCCGTAGGC-1 315
#> b_TTTGTCATCCTCGCAT-1 208
#> b_TTTGTCATCGCCGTGA-1 287
#> b_TTTGTCATCGTTTGCC-1 358
#> Name: metacell, Length: 29581, dtype: int32>The following code chunk adds a columns (named membership) containing the single_cell assignments to the obs attribute in the single-cell anndata object.
The membership information is required to compute metacells quality metrics as shown in chapter 2.
# make a membership -- index of metacells to which single cells belong to
ad.obs['membership'] = [int(i)+1 if i >= 0 else np.nan for i in ad.obs.metacell] Retrieve aggregated metacell data
The function mc.pl.collect_metacells() should be used to subsequently retrieve an anndata object containing the data at the metacells level instead of the single-cell level.
This function will store in the X data matrix of the anndata object a matrix of gene fraction (i.e., the sum of all gene levels in a metacell sums to 1)
and it will store the total UMIs per gene per metacell in the layer total_umis.
mc_ad = mc.pl.collect_metacells(ad, name='metacells', random_seed = 123456)
#> set metacells.obs[grouped]: 401 int64s
#> set metacells.obs[total_umis]: 401 float64s
#> set metacells.layers[total_umis]: ndarray 401 X 16992 float32s
#> set metacells.obs[__zeros_downsample_umis]: 401 int64s
#> set metacells.layers[zeros]: ndarray 401 X 16992 int32s
#> set bmcite.clean.obs[metacell_name]: 29581 <U8s
#> set metacells.var[genes]: 16992 objects
#> set metacells.var[bursty_lonely_gene]: 0 true (0%) out of 16992 bools
#> set metacells.var[properly_sampled_gene]: 16992 true (100%) out of 16992 bools
#> set metacells.var[excluded_gene]: 0 true (0%) out of 16992 bools
#> set metacells.var[full_gene_index]: 16992 int32s
#> set metacells.var[lateral_gene]: 245 true (1.442%) out of 16992 bools
#> set metacells.var[noisy_gene]: 20 true (0.1177%) out of 16992 bools
#> set metacells.var[selected_gene]: 1807 true (10.63%) out of 16992 bools
#> set metacells.var[rare_gene]: 10 true (0.05885%) out of 16992 bools
#> set metacells.var[rare_gene_module]: 16992 int32s
#> set metacells.obs[metacells_rare_gene_module]: 401 int32s
#> set metacells.obs[rare_metacell]: 1 true (0.2494%) out of 401 bools
#> set metacells.uns[outliers]: 5
#> set metacells.uns[metacells_algorithm]: metacells.0.9.0
mc_ad.shape
#> (401, 16992)
mc_ad.X.sum(axis=1)[:5]
#> matrix([[1.],
#> [1.],
#> [1.],
#> [1.],
#> [1.]], dtype=float32)
mc_ad.layers['total_umis']
#> array([[ 2., 1., 0., ..., 0., 0., 0.],
#> [ 1., 0., 0., ..., 0., 0., 0.],
#> [ 5., 1., 0., ..., 0., 0., 0.],
#> ...,
#> [ 5., 0., 0., ..., 0., 0., 0.],
#> [ 2., 2., 0., ..., 0., 0., 0.],
#> [10., 1., 0., ..., 0., 0., 0.]], dtype=float32)1.2.5 Annotate metacells (using available annotations)
If single-cell annotations are available in the original single-cell anndata object. We can transfer these annotations to the metacell anndata object
using the mc.tl.convey_obs_to_group() function which will associate each metacell to the most frequent annotation (categorical) or
averaged annotation (continuous) across the single-cells composing the metacell
(use of the mc.ut.most_frequent and np.mean respectively in the mode paratemer).
# Assign a single value for each metacell based on the cells.
mc.tl.convey_obs_to_group(
adata=ad, gdata=mc_ad,
property_name=annotation_label, to_property_name=annotation_label,
method=mc.ut.most_frequent # This is the default, for categorical data
)
#> set metacells.obs[celltype_simplified]: 401 <U18s
# Compute the fraction of cells with each possible value in each metacell:
mc.tl.convey_obs_fractions_to_group(
adata=ad, gdata=mc_ad,
property_name=annotation_label, to_property_name=annotation_label
)
#> set metacells.obs[celltype_simplified_fraction_of_B cell]: 401 float64s
#> set metacells.obs[celltype_simplified_fraction_of_CD14 Mono]: 401 float64s
#> set metacells.obs[celltype_simplified_fraction_of_CD16 Mono]: 401 float64s
#> set metacells.obs[celltype_simplified_fraction_of_GMP]: 401 float64s
#> set metacells.obs[celltype_simplified_fraction_of_HSC]: 401 float64s
#> set metacells.obs[celltype_simplified_fraction_of_LMPP]: 401 float64s
#> set metacells.obs[celltype_simplified_fraction_of_MEP]: 401 float64s
#> set metacells.obs[celltype_simplified_fraction_of_NK]: 401 float64s
#> set metacells.obs[celltype_simplified_fraction_of_Naive T cell]: 401 float64s
#> set metacells.obs[celltype_simplified_fraction_of_Non-Naive CD4 cell]: 401 float64s
#> set metacells.obs[celltype_simplified_fraction_of_Non-Naive CD8 cell]: 401 float64s
#> set metacells.obs[celltype_simplified_fraction_of_Plasmablast]: 401 float64s
#> set metacells.obs[celltype_simplified_fraction_of_Prog_B]: 401 float64s
#> set metacells.obs[celltype_simplified_fraction_of_Prog_DC]: 401 float64s
#> set metacells.obs[celltype_simplified_fraction_of_Unconventional T]: 401 float64s
#> set metacells.obs[celltype_simplified_fraction_of_cDC2]: 401 float64s
#> set metacells.obs[celltype_simplified_fraction_of_pDC]: 401 float64s1.2.6 Save output
For future downstream analyses in python (section 3.2), we save the metacell counts in an Anndata object:
## Save single-cell metadata (i.e., `raw.obs` dataframe) in the metacell adata object
mc_ad.uns = ad.uns.copy()
mc_ad.uns['sc.obs'] = ad.obs.copy()
# save the requested gamma
mc_ad.uns['gamma'] = gamma
# save metacell size
mc_ad.obs.rename(columns={'grouped':'size'}, inplace = True)
print("Saving metacell object for the "+ proj_name+ " dataset using "+ MC_tool)
#> Saving metacell object for the bmcite dataset using MC2
mc_ad.write_h5ad(os.path.join('./data', proj_name, f'metacell_{MC_tool}.h5ad'))For future QCs and downstream analyses in R (section 3.1), we save the metacell counts in a Seurat object:
library(Seurat)
# If you have Seurat V5 installed, specify that you want to analyze Seurat V4 objects
if(packageVersion("Seurat") >= 5) {options(Seurat.object.assay.version = "v4"); print("you are using seurat v5 with assay option v4")}
library(anndata)
library(reticulate)
adata_mc <- read_h5ad(paste0("data/", py$proj_name, "/metacell_MC2.h5ad"))
# Save counts and metadata in a Seurat object
countMatrix <- Matrix::t(adata_mc$X)
colnames(countMatrix) <- adata_mc$obs_names
rownames(countMatrix) <- adata_mc$var_names
MC.seurat <- CreateSeuratObject(counts = as(countMatrix, 'CsparseMatrix'), meta.data = as.data.frame(adata_mc$obs))
#> Warning: Invalid name supplied, making object name syntactically valid. New
#> object name is
#> sizetotal_umisX__zeros_downsample_umismetacells_rare_gene_modulerare_metacellcelltype_simplifiedcelltype_simplified_fraction_of_B.cellcelltype_simplified_fraction_of_CD14.Monocelltype_simplified_fraction_of_CD16.Monocelltype_simplified_fraction_of_GMPcelltype_simplified_fraction_of_HSCcelltype_simplified_fraction_of_LMPPcelltype_simplified_fraction_of_MEPcelltype_simplified_fraction_of_NKcelltype_simplified_fraction_of_Naive.T.cellcelltype_simplified_fraction_of_Non.Naive.CD4.cellcelltype_simplified_fraction_of_Non.Naive.CD8.cellcelltype_simplified_fraction_of_Plasmablastcelltype_simplified_fraction_of_Prog_Bcelltype_simplified_fraction_of_Prog_DCcelltype_simplified_fraction_of_Unconventional.Tcelltype_simplified_fraction_of_cDC2celltype_simplified_fraction_of_pDC;
#> see ?make.names for more details on syntax validity
MC.seurat@misc[["var_features"]] <- rownames(adata_mc$var)[which(adata_mc$var$selected_gene == T)]
# Save membership in misc
MC.seurat@misc$cell_membership <- py$ad$obs['membership']
if(packageVersion("Seurat") >= 5) {
MC.seurat[["RNA"]] <- as(object = MC.seurat[["RNA"]], Class = "Assay")
}
saveRDS(MC.seurat, file = paste0('./data/', py$proj_name, '/metacell_MC2.rds'))1.3 SEACells (Python)
In this section, we construct metacells using SEACells.
1.3.1 Method
The SEAcells method builds a single-cell kNN graph from the Euclidean distance in the principal component space (SVD for scATAC-seq) space. Distances in the graph are transformed to affinity by applying an adaptive Gaussian kernel. The affinity matrix is then decomposed into archetypes (linear combination of cells) and membership matrices (cells as a linear combination of archetypes). Single cells are assigned to a given metacell based on the maximum membership value of the corresponding archetype.
The code provided in this section is adapted from the author’s jupyter notebook. For more information on the method, please refer to our review (Review?) and the original paper (Persad et al. 2023).
Importing python packages
To run the SEACells, the following python packages need to be imported:
import os
import pandas as pd
import scanpy as sc
import SEACells
import randomIf you don’t have these packages installed, please refer to the section 0.1.
1.3.2 Data loading
Similarly to SuperCell and MC2, we will run SEACells on the single-cell dataset composed of around 30’000 bone marrow cells. Please follow the section 0.2 to retrieve these data from the SeuratData package and save the data in the following file: “data/bmcite/singlecell_anndata_filtered.h5ad”.
MC_tool = "SEACells"
proj_name = "bmcite"
annotation_label = 'celltype_simplified'
ad = sc.read(os.path.join("data", proj_name, "singlecell_anndata_filtered.h5ad"))
ad.var.index = ad.var.genes
ad = sc.pp.subsample(ad, n_obs=10000, copy=True)Note that since SEACells can take a substential amount of time to run we downsampled the data to 10’000 cells, you can comment the last line of the previous code chunk to run SEACells on all the cells.
1.3.3 Filtering steps
In this tutorial, the data have been pre-filterd and SEACells does not perform additionnal filtering.
1.3.4 Building metacells
Metacells construction using SEACells requires 2 main inputs: i) an anndata object (build_kernel_on parameter), and
ii) a key indicating which matrix in the obsm attribute of the anndata object should be considered to compute the kernel needed for archetypal analysis (build_kernel_on parameter).
Important optional inputs are: the number of metacells to identify (n_SEACells parameter), which is used as input of the archetypal analysis,
ii) the number of neighbors to consider for the knn graph (n_neighbors parameter).
Data pre-processing
The following code chunk saves the raw counts of the filtered data in the raw attribute of the anndata object. The raw counts will be used later for metacells aggregation.
raw_ad = sc.AnnData(ad.X)
raw_ad.obs_names, raw_ad.var_names = ad.obs_names, ad.var_names
ad.raw = raw_adTo build the kernel for archetypal analysis, SEACells requires a lower-dimensionnal embedding of the data (for example using PCA for scRNA-Seq data or SVD for scATAC-Seq data).
In the next code chunk, we follow standard pre-processing steps prior to PCA computation, i.e., data normalization, log transformation, identification of highly variable genes.
PCA components are saved in the obsm attribute of the anndata object.
# Normalize cells, log transform and compute highly variable genes
sc.pp.normalize_per_cell(ad, 10000)
sc.pp.log1p(ad)
sc.pp.highly_variable_genes(ad, n_top_genes=2000)
# Compute principal components
n_comp = 30
sc.tl.pca(ad, n_comps=n_comp, use_highly_variable=True)
# Run UMAP for visualization
sc.pp.neighbors(ad)
sc.tl.umap(ad)Setting up SEACells parameters
In this tutorial, we will use in the SEACells model the 30 first principal components resulting from the PCA to build the knn graph which will be used to compute the kernel.
The number of neighbors to considered for the knn graph can be fixed using the n_neighbors parameter (here 15).
As mentioned previously, users should provide as input the number of metacells required (n_SEACells parameter). This number can be defined as the ratio between the number of single cells and the desired graining level (gamma parameter in the following code chunk).
In this example, we choose a graining level of 50.
build_kernel_on = 'X_pca' # key in ad.obsm to use for computing metacells
n_waypoint_eigs = 10 # Number of eigenvalues to consider when initializing metacells
n_neighbors = 15 # Number of neighbors used for graph construction
gamma = 50 # the requested graining level
n_SEACells = int(ad.shape[0]/gamma) # the requested number of metacells Initializing the SEACells model
The SEACells model is initialized with the previously defined parameters using the SEACells.core.SEACells function.
model = SEACells.core.SEACells(ad,
build_kernel_on = build_kernel_on,
n_SEACells = n_SEACells,
n_waypoint_eigs = n_waypoint_eigs,
n_neighbors = n_neighbors,
convergence_epsilon = 1e-3,
verbose = True)
#> Welcome to SEACells!Kernel computation is performed using the mconstruct_kernel_matrix function.
model.construct_kernel_matrix()
#> Computing kNN graph using scanpy NN ...
#> Computing radius for adaptive bandwidth kernel...
#> 0%| | 0/10000 [00:00<?, ?it/s]
#> Making graph symmetric...
#> Parameter graph_construction = union being used to build KNN graph...
#> Computing RBF kernel...
#> 0%| | 0/10000 [00:00<?, ?it/s]
#> Building similarity LIL matrix...
#> 0%| | 0/10000 [00:00<?, ?it/s]
#> Constructing CSR matrix...
#>
#> /opt/conda/envs/MetacellAnalysisToolkit/lib/python3.9/site-packages/scipy/sparse/_index.py:143: SparseEfficiencyWarning: Changing the sparsity structure of a csr_matrix is expensive. lil_matrix is more efficient.
#> self._set_arrayXarray(i, j, x)
M = model.kernel_matrixMetacells are initialized using the initialize_archetypes function.
The SEACells archetypes initialization is based on cells sampling and thus is stochastic. User can fix a seed for reproducible results.
To check that the archetypes are evenly spread, users can visualize them using the plot.plot_initialization function.
# set seed for reproducibility
# random.seed(123)
# Initialize archetypes
model.initialize_archetypes()
#> Building kernel on X_pca
#> Computing diffusion components from X_pca for waypoint initialization ...
#> Done.
#> Sampling waypoints ...
#> Done.
#> Selecting 184 cells from waypoint initialization.
#> Initializing residual matrix using greedy column selection
#> Initializing f and g...
#> Selecting 16 cells from greedy initialization.
#>
#>
0%| | 0/26 [00:00<?, ?it/s]
38%|###8 | 10/26 [00:00<00:00, 92.87it/s]
77%|#######6 | 20/26 [00:00<00:00, 90.85it/s]
100%|##########| 26/26 [00:00<00:00, 92.20it/s]
# Visualize the initialization
SEACells.plot.plot_initialization(ad, model, plot_basis='X_umap') 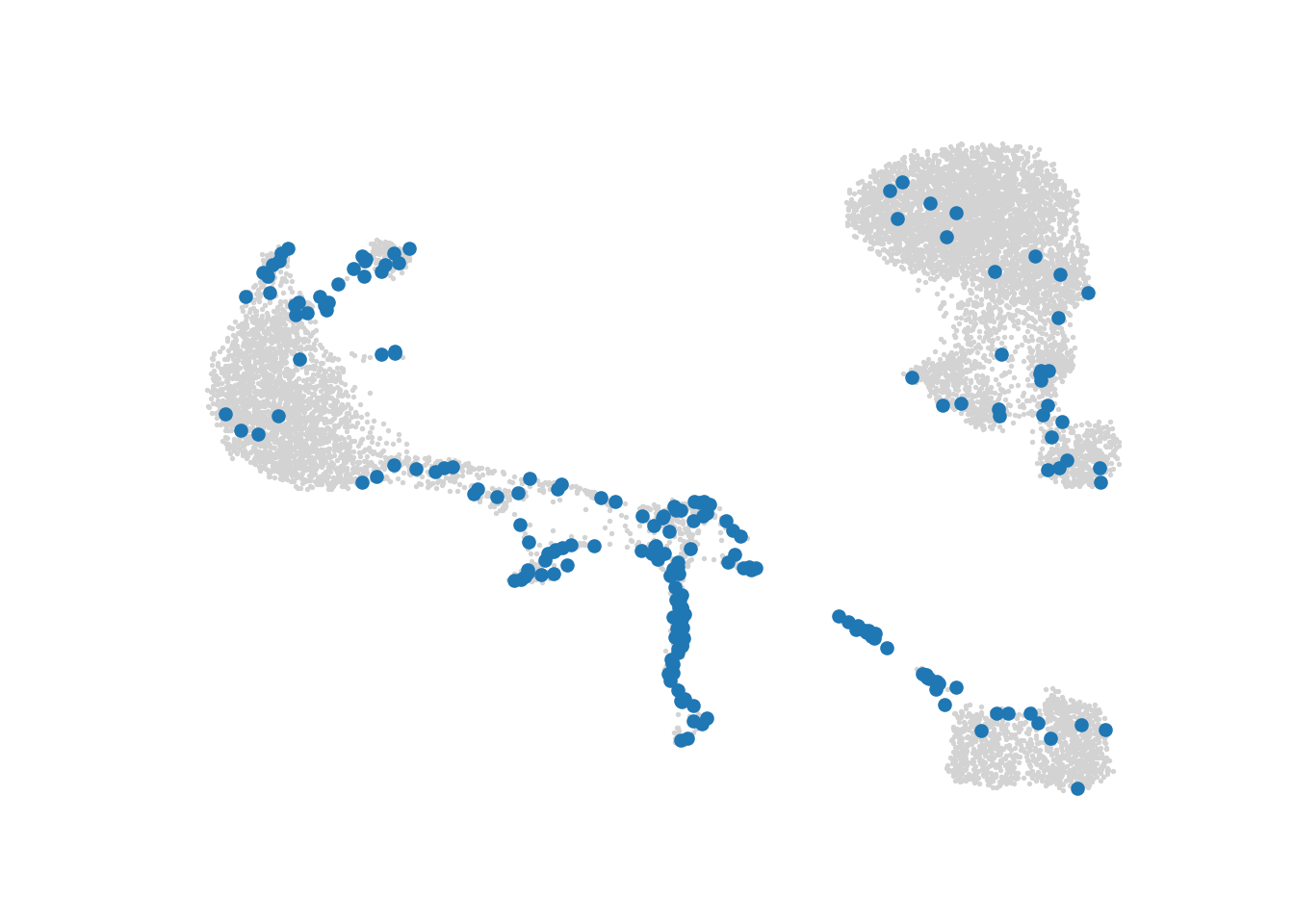
Fitting the SEACells model to identify metacells
The identification of the archetypes is an iterative process. In this example, we fixed the minimum and maximum number of iteration to 10 and 50 respectively.
We then check the model convergence using the plot_convergence function.
model.fit(min_iter = 10, max_iter = 100)
#> Randomly initialized A matrix.
#> Setting convergence threshold at 0.17973
#> Starting iteration 1.
#> Completed iteration 1.
#> Starting iteration 10.
#> Completed iteration 10.
#> Converged after 11 iterations.
model.plot_convergence()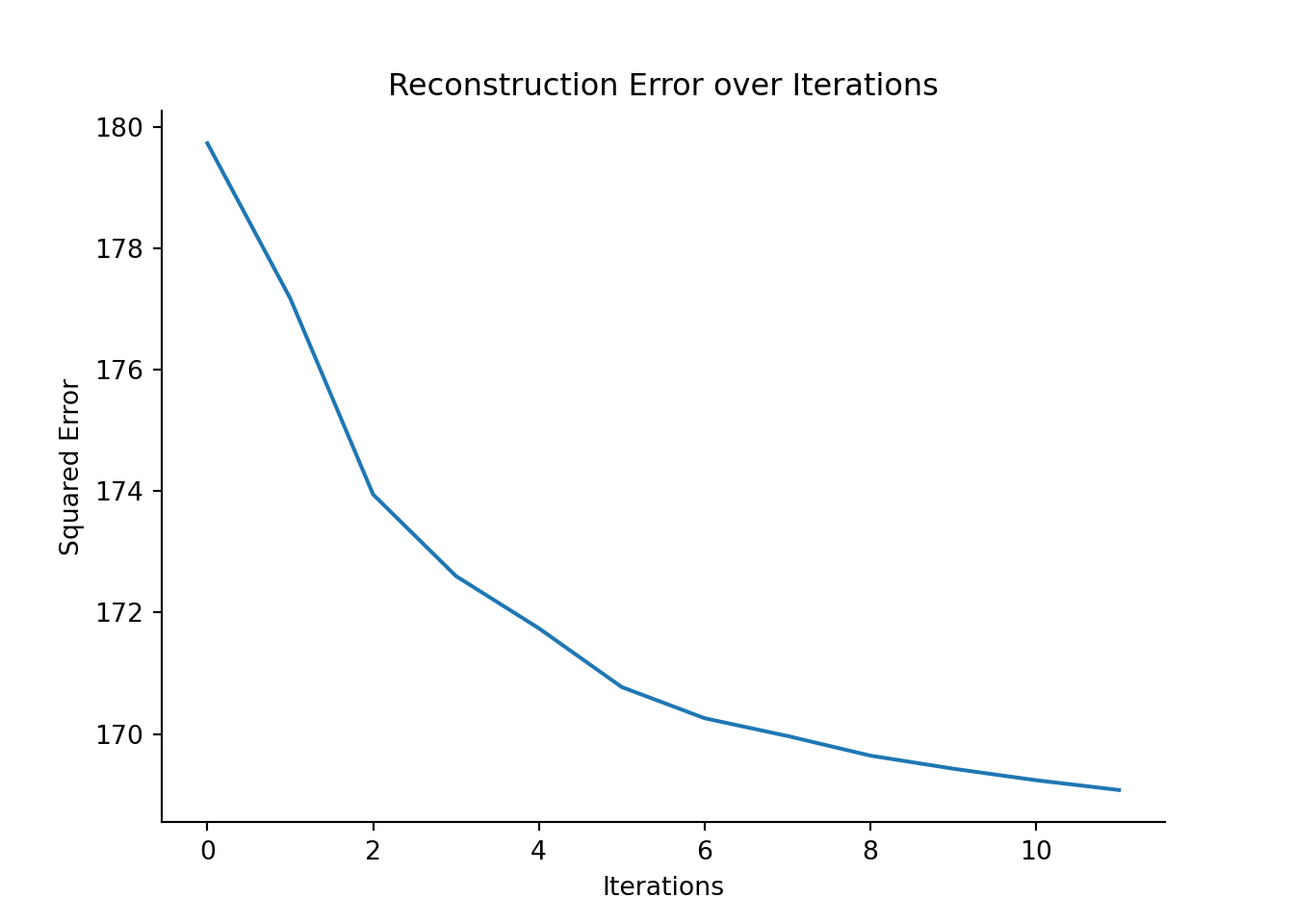
Once the final archetypes have been identified, we can assign each single-cell to one metacell (hard assignments).
These assignments (membership) can be retrieved using the get_hard_assignments function or extracted from the anndata object using ad.obs["SEACell"].
In this tutorial, we will only consider hard assignments. However, the SEACells package also provides the option to retrieve soft assignments (multiple weighted assignments for each cell) using the get_soft_assignments function.
For more details on the soft assignments, please refer to the SEACell paper and the original author’s jupyter notebook.
membership = model.get_hard_assignments()
membership.head
#> <bound method NDFrame.head of SEACell
#> index
#> b_TGACTTTGTGTTTGGT-1 SEACell-16
#> a_TTCTCCTTCCGCGCAA-1 SEACell-35
#> a_ACTATCTTCTTGAGAC-1 SEACell-134
#> b_GTTCTCGAGTAACCCT-1 SEACell-112
#> b_ACCCACTGTGGGTCAA-1 SEACell-20
#> ... ...
#> b_AGGGTGATCGCATGAT-1 SEACell-179
#> b_TCATTTGTCATATCGG-1 SEACell-142
#> a_ATCGAGTGTAGCGCTC-1 SEACell-43
#> a_CCACGGAAGAATCTCC-1 SEACell-25
#> b_TGTCCCATCCATGAAC-1 SEACell-171
#>
#> [10000 rows x 1 columns]>
ad.obs["SEACell"].head
#> <bound method NDFrame.head of index
#> b_TGACTTTGTGTTTGGT-1 SEACell-16
#> a_TTCTCCTTCCGCGCAA-1 SEACell-35
#> a_ACTATCTTCTTGAGAC-1 SEACell-134
#> b_GTTCTCGAGTAACCCT-1 SEACell-112
#> b_ACCCACTGTGGGTCAA-1 SEACell-20
#> ...
#> b_AGGGTGATCGCATGAT-1 SEACell-179
#> b_TCATTTGTCATATCGG-1 SEACell-142
#> a_ATCGAGTGTAGCGCTC-1 SEACell-43
#> a_CCACGGAAGAATCTCC-1 SEACell-25
#> b_TGTCCCATCCATGAAC-1 SEACell-171
#> Name: SEACell, Length: 10000, dtype: object>Retrieve aggregated metacell data
The core.summarize_by_SEACell function can be used to generate a metacell count matrix (aggregation of counts across all cells belonging to each metacell).
mc_ad = SEACells.core.summarize_by_SEACell(ad, SEACells_label='SEACell', summarize_layer='raw', celltype_label=annotation_label)
#>
0%| | 0/200 [00:00<?, ?it/s]
2%|2 | 5/200 [00:00<00:04, 41.08it/s]
5%|5 | 10/200 [00:00<00:04, 42.65it/s]
8%|7 | 15/200 [00:00<00:04, 43.14it/s]
10%|# | 20/200 [00:00<00:04, 43.80it/s]
12%|#2 | 25/200 [00:00<00:04, 43.63it/s]
15%|#5 | 30/200 [00:00<00:03, 44.28it/s]
18%|#7 | 35/200 [00:00<00:03, 43.78it/s]
20%|## | 40/200 [00:00<00:03, 44.36it/s]
22%|##2 | 45/200 [00:01<00:03, 44.19it/s]
25%|##5 | 50/200 [00:01<00:03, 43.67it/s]
28%|##7 | 55/200 [00:01<00:03, 44.14it/s]
30%|### | 60/200 [00:01<00:03, 44.71it/s]
32%|###2 | 65/200 [00:01<00:03, 44.34it/s]
35%|###5 | 70/200 [00:01<00:02, 44.62it/s]
38%|###7 | 75/200 [00:01<00:02, 45.00it/s]
40%|#### | 80/200 [00:01<00:02, 45.14it/s]
42%|####2 | 85/200 [00:01<00:02, 45.70it/s]
45%|####5 | 90/200 [00:02<00:02, 45.57it/s]
48%|####7 | 95/200 [00:02<00:02, 45.68it/s]
50%|##### | 100/200 [00:02<00:02, 45.93it/s]
52%|#####2 | 105/200 [00:02<00:02, 46.01it/s]
55%|#####5 | 110/200 [00:02<00:01, 45.38it/s]
57%|#####7 | 115/200 [00:02<00:01, 45.28it/s]
60%|###### | 120/200 [00:02<00:01, 45.17it/s]
62%|######2 | 125/200 [00:02<00:01, 45.62it/s]
65%|######5 | 130/200 [00:02<00:01, 46.11it/s]
68%|######7 | 135/200 [00:03<00:01, 46.18it/s]
70%|####### | 140/200 [00:03<00:01, 46.45it/s]
72%|#######2 | 145/200 [00:03<00:01, 46.97it/s]
75%|#######5 | 150/200 [00:03<00:01, 46.77it/s]
78%|#######7 | 155/200 [00:03<00:00, 47.49it/s]
80%|######## | 160/200 [00:03<00:00, 47.37it/s]
82%|########2 | 165/200 [00:03<00:00, 47.35it/s]
85%|########5 | 170/200 [00:03<00:00, 47.54it/s]
88%|########7 | 175/200 [00:03<00:00, 47.55it/s]
90%|######### | 180/200 [00:03<00:00, 47.90it/s]
92%|#########2| 185/200 [00:04<00:00, 48.28it/s]
95%|#########5| 190/200 [00:04<00:00, 48.68it/s]
98%|#########7| 195/200 [00:04<00:00, 49.00it/s]
100%|##########| 200/200 [00:04<00:00, 49.23it/s]
100%|##########| 200/200 [00:04<00:00, 45.95it/s]1.3.5 Visualize metacells
To visualize the metacells, we can project the metacells on the single-cell UMAP representation using the plot.plot_2D included in the SEACells package.
SEACells.plot.plot_2D(ad, key='X_umap', colour_metacells=True)
Save output
For future downstream analyses in python (section 3.2), we save the metacell counts in an Anndata object:
print("Saving metacell object for the "+ proj_name+ " dataset using "+ MC_tool)
#> Saving metacell object for the bmcite dataset using SEACells
# Save metacell sizes
label_df = ad.obs[['SEACell']].reset_index()
mc_ad.obs = mc_ad.obs.join(pd.DataFrame(label_df.groupby('SEACell').count().iloc[:, 0]).rename(columns={'index':'size'}))
# save pca used to compute metacells
mc_ad.uns['var_features']=ad.var_names[ad.var.highly_variable].tolist()
mc_ad.uns['sc.pca']=ad.obsm['X_pca']
mc_ad.uns['sc.umap']=ad.obsm['X_umap']
mc_ad.write_h5ad(os.path.join('./data', proj_name, f'metacell_{MC_tool}.h5ad'))For future downstream analyses in R (section 3.1), we save the metacell counts in a Seurat object:
library(Seurat)
library(Seurat)
# If you have Seurat V5 installed, specify that you want to analyze Seurat V4 objects
if(packageVersion("Seurat") >= 5) {options(Seurat.object.assay.version = "v4"); print("you are using seurat v5 with assay option v4")}
library(anndata)
library(reticulate)
adata_mc <- read_h5ad(paste0("data/", py$proj_name, "/metacell_SEACells.h5ad"))
# Save counts and metadata in a Seurat object
countMatrix <- Matrix::t(adata_mc$X)
colnames(countMatrix) <- adata_mc$obs_names
rownames(countMatrix) <- adata_mc$var_names
MC.seurat <- CreateSeuratObject(counts = as(countMatrix, 'CsparseMatrix'), meta.data = as.data.frame(adata_mc$obs))
# MC.seurat@misc[["sc.pca"]] <- adata_mc$uns$sc.pca
# MC.seurat@misc[["sc.umap"]] <- adata_mc$uns$sc.umap
MC.seurat@misc[["var_features"]] <- adata_mc$uns$var_features
pca.res <- adata_mc$uns$sc.pca
rownames(pca.res) <- rownames(py$ad$obs)
MC.seurat@misc$sc.pca <- CreateDimReducObject(
embeddings = pca.res,
key = "PC_",
assay = "RNA"
)
#> Warning: No columnames present in cell embeddings, setting to 'PC_1:30'
# Save membership in misc
MC.seurat@misc$cell_membership <- data.frame(row.names = rownames(py$membership), membership = py$membership$SEACell)
if(packageVersion("Seurat") >= 5) {
MC.seurat[["RNA"]] <- as(object = MC.seurat[["RNA"]], Class = "Assay")
}
saveRDS(MC.seurat, file = paste0('./data/', py$proj_name, '/metacell_SEACells.rds'))